新日本科学PPDのサービス
Our Work
新薬開発の包括的なサービス
医薬品として承認されるまでには数多くの段階があり、膨大な時間と労力を必要とします。そこで、私たちは開発効率を高め、
より早く新しい薬を生み出すため、臨床試験・製造販売後臨床試験のさまざまな業務を代行・サポートしています。
モニタリングから集積されたデータの処理・解析、規制当局に提出する申請書類の作成に至るまで、
臨床開発のスペシャリストとして最高品質のサービスを提供します。
また、遺伝子治療に関する国際共同臨床試験を支援するために「遺伝子組み換え生物等の使用等の規制による生物の多様性の確保に関する法律」に基づく「第一種使用規程承認申請」の手続き業務を完了した実績もあります。
サービスの詳細は、PPD Globalのウェブサイトよりお問い合わせ下さい。
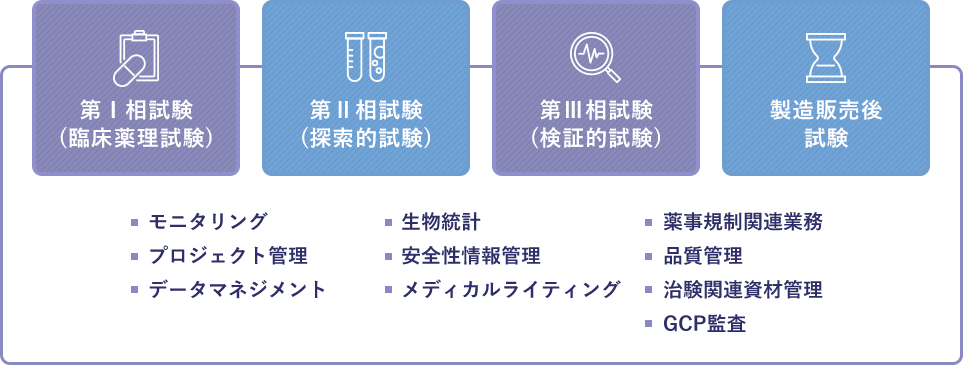
新薬開発のプロセス
医薬品が安全に安心して使用できるものであると国の基準を満たすまでには、数々の試験段階があります。
新日本科学PPDでは、臨床試験から製造販売後調査までのそれぞれの試験段階において、様々な部署が協働し、
スペシャリストによる包括的なサポートを実現します。
医療機関・治験責任医師選定

- ・受託内容の確認
- ・事前調査 (フィジビリティ調査)、打診
治験開始に先立ち、治験依頼者の要望を満たす医療機関・治験責任医師の選定を行います。
その医療機関で治験を実施するのに、十分な設備・人員を有していることや、緊急時の被験者の安全を確保できる体制が整っていることなどを事前に確認し、安全に治験を行う体制を整えます。
メディカルモニタリング
- ・治験実施計画書のトレーニング
- ・対象疾患のトレーニング
医師免許を有するMedical Monitorにより、モニタリングチームへ治験実施計画書や対象疾患の理解を深めさせることで、より専門性の高いモニタリングを実施可能とします。
治験届前準備 治験届提出
- ・治験実施の科学的妥当性や治験デザインに関する助言
- ・治験開始に必要な薬事規制に対する対応(特に、再生医療等製品)
- ・承認申請までに必要な臨床・非臨床データに関する助言(薬事戦略)
- ・治験計画届書の提出(照会事項対応含む)
- ・治験計画変更届書、治験中止届書、治験終了届書の提出
- ・治験届に関する疑義事項の対応(PMDA等との交渉含む)
- ・最新の国内薬事規制(新規ICHガイドライン含む)に関する情報の収集
医薬品医療機器法において、保健衛生上の見地から治験の実態を把握し、治験の安全性を確保するため、治験依頼者(製薬企業等)及び医師又は歯科医師(自ら治験を実施する者)は、厚生労働大臣への治験計画の届け出が義務づけられています。治験計画の届出は、PMDAに対し提出され、その届出時期・届出内容などが規定されています。
契約締結、治験開始前準備

- ・必要な資料作成 (IRB申請資料、契約書・覚書等)
- ・治験開始までの進捗状況確認 (各種システム等)
- ・契約管理業務
- ・クライアントである製薬企業等との交渉
- ・医療機関側のあらゆる運営・品質面の管理
- ・プロジェクトチームのパフォーマンス管理
- ・リスクマネジメント


- ・文書の確認、管理
- ・医療機関/治験責任医師への安全性情報の発送
- ・印刷/翻訳等の各種ベンダー対応
- ・医療機関からの請求書対応
- ・監査時におけるファイルの準備やアシスト
モニタリング

- ・医療機関への訪問・情報伝達
- ・症例登録促進/進捗管理
- ・モニタリング報告書の作成
- ・必須文書の保管状況の確認
- ・SDV(被験者さまのデータ確認)
- ・症例報告書の回収・点検
CRAはBlinded CRA (盲検CRA)とUnblinded CRA(非盲検CRA)の二つに分けられ、モニタリング業務を遂行しています。多くを占めるのがBlinded CRAですが、Unblinded CRAも臨床試験を円滑に進めるにあたって重要な役割を担っています。
Unblinded CRAは治験の盲検性を保つために、治験薬の特定や管理・データのモニタリングをBlinded CRAとは別に行っています。二重盲検試験とは、治験薬と対照薬のどの薬剤を服用しているのか、被験者や医療機関にも明らかにせず行う臨床試験のことです。
治験の盲検性を維持することで、試験結果の偏りを無くし、正確なデータを入手するために重要な役割を果たします。
- ・安全性情報の集積・分析・評価
- ・安全対策検討・決定
- ・安全性情報の提供
- ・厚生労働省(PMDA)への安全性情報の報告
- ・グローバル/国内治験におけるメディカルモニタリング業務
- ・治験国内管理人安全性情報のメディカルレビュー
- ・試験領域の社内トレーニング
- ・治験関連文書の正確性確認
- ・治験の手続きが正しく実施されているかの確認
- ・文書保管・管理
- ・手順書の管理
- ・文書保管
- ・問題発生時の原因究明・再発防止策検討
- ・規制・手順書に関する社内問い合わせ窓口
- ・品質管理体制の維持

データマネジメント、統計解析、メディカルライティング
- ・GCP、治験実施計画書、業務手順書を遵守して治験が実施されているかの評価
- ・治験に関連する各種コンプライアンス遵守状況の評価
- ・治験薬の回収
- ・症例報告書の回収
- ・治験終了報告書の作成補助
- ・治験終了通知書を医療機関より入手
DM・統計解析・メディカルライティング部門
- ・治験データの固定
- ・結果報告書、承認申請に必要な文書の作成

治験終了手続き後に規制当局・依頼者に収集したデータを提出し、治験が終了となります。そのため、治験の終了時には、医療機関・PI選定から治験終了までの全てのデータが完全でなければなりません。医療機関担当CRAはもちろん、その他の該当治験に携わった部署が連携をとり、データの整合性を確認します。
ダミー6
製造販売後臨床試験
- ・新たな作用・副作用に関する情報収集
- ・製造販売調査の進捗管理
- ・安全性情報の管理
治験が終了し、無事に承認申請を取得できたとしても、患者さまに医薬品を安心して使用して頂くために製造販売後調査が設けられています。
医薬品としての有効性・安全性の確認と、販売前の治験で得られなかった新たな作用・副作用に関する情報収集を主に行います。販売前の臨床試験とは異なる法律(GPSP, GVP)に従い、調査を行うため専門の知識を持ったスタッフが行います。
